
Tabella dei contenuti
Curarsi
Quali sono i trattamenti per l’emofilia?
Essendo l'emofilia un'alterazione della coagulazione del sangue, l'attuale terapia standard consiste nel somministrare il fattore della coagulazione mancante mediante un'iniezione.
Panoramica delle terapie contro l’emofilia
Oggi, per i pazienti emofilici sono disponibili le seguenti terapie:
| Approccio terapeutico | Somministrazione | |
|---|---|---|
| Emofilia A (fattore VIII mancante o carente) | Preparati contenenti i fattori di coagulazione (sostituzione del fattore VIII) | Endovenosa (iniezione nella vena del braccio) |
| Anticorpo bispecifico(assume la funzione del fattore VIII) | Sottocutanea (iniezione direttamente sotto la pelle) | |
| Emofilia B (fattore IX mancante o carente) | Preparati contenenti i fattori della coagulazione (sostituzione del fattore IX) | Endovenosa (iniezione nella vena del braccio) |
Base del trattamento: sostituzione del fattore della coagulazione mancante
Da quando è noto che l’emofilia comporta una parziale o totale mancanza di un fattori della coagulazione, che impedisce al sangue di coagulare correttamente in caso di lesione, si è affermata la terapia standard attuale: il fattore della coagulazione viene sostituito tramite un preparato contenente i fattori della coagulazione o un anticorpo. Nell’emofilia A è interessato il fattore VIII, mentre nell’emofilia B il fattore IX.
Negli anni ’50 del Novecento, all’inizio della cosiddetta terapia sostitutiva dei fattori della coagulazione, il fattore mancante veniva ricavato insieme ad altre proteine dal sangue umano di un donatore. Questa miscela di proteine, chiamata frazione di Cohn, veniva somministrata al paziente emofilico mediante iniezione endovenosa in caso di sanguinamento acuto. Ciò consentiva di completare la cascata della coagulazione, arrestando i sanguinamenti in maniera sicura.
Alcuni anni più tardi, alcuni ricercatori riuscirono a ricavare dal plasma sanguigno il singolo fattore della coagulazione adatto per ogni specifica alterazione: fu così possibile somministrare ai pazienti emofilici solo il fattore effettivamente mancante, ossia il fattore VIII o il fattore IX. Il nuovo preparato venne definito concentrato plasmatico del fattore della coagulazione. Da più di 10 anni sono presenti sul mercato preparati dei fattori della coagulazione sia plasmatici che ricombinanti. La maggior parte dei pazienti utilizza i preparati a domicilio (autoinfusione) e non deve più recarsi regolarmente in ospedale.
Ricerca e sviluppo nel campo dell'emofilia
Il medicamento contenente il fattore della coagulazione veniva ricavato inizialmente da sangue donato. All’inizio era ancora possibile, quindi, che il donatore potesse trasmettere al paziente non solo il fattore della coagulazione, ma anche agenti patogeni. Di conseguenza, numerosi pazienti emofilici contrassero l’epatite B e C. Negli anni ’80 del Novecento la situazione fu particolarmente drammatica, poiché con i medicamenti contenenti i fattori della coagulazione venne trasmesso l’HIV.
Si cercarono pertanto nuove metodologie per rendere più sicuri questi medicamenti. Per questo motivo, da allora si utilizzano procedure speciali per distruggere o disattivare eventuali agenti patogeni (virus e batteri) nei medicamenti (plasmatici) prodotti da plasma sanguigno. L’ulteriore ricerca ha reso possibile la produzione dei fattori della coagulazione con biotecnologie, senza l’impiego di sangue umano: dal 2004 sono disponibili in Svizzera medicamenti prodotti esclusivamente con biotecnologie, i cosiddetti medicamenti ricombinanti.
Dal 2019 è disponibile un preparato sottocutaneo per il trattamento profilattico dell’emofilia A grave. Un anticorpo bispecifico, prodotto con biotecnologie, assume nel corpo la funzione del fattore VIII mancante o carente e accelera la coagulazione del sangue. Il preparato viene iniettato direttamente sotto la pelle (iniezione sottocutanea).
Profilassi o trattamento a richiesta: il fattore discriminante è il grado di gravità
I fattori della coagulazione mancanti non vengono somministrati solo per trattamenti acuti, ma, in base al grado di gravità dell’emofilia, anche a scopo preventivo, ossia per trattamenti profilattici. La profilassi offre benefici soprattutto ai pazienti con emofilia moderata o severa, riducendo i sanguinamenti spontanei e i danni articolari a lungo termine.
Oggi, molti pazienti possono quindi condurre una vita con poche limitazioni. Tuttavia, occorre tenere presente che i preparati contenenti fattori della coagulazione devono essere somministrati con una certa regolarità. Ciò dipende dal fatto che i fattori della coagulazione vengono metabolizzati nel fegato. L’emivita, ovvero la velocità con cui si riduce la concentrazione di un certo fattore, varia da individuo a individuo e da preparato a preparato. Per calcolare la frequenza di somministrazione di un preparato, è necessario stabilire l’emivita del fattore nel sangue.
L’emivita indica precisamente il tempo impiegato dalla quantità del fattore per dimezzarsi nel sangue. Se l’emivita è di dodici ore, significa che dopo dodici ore sarà presente solo metà della quantità iniziale del fattore. Dopo altre dodici ore, sarà rimasta la metà della metà, e così via. Dopo un certo periodo di tempo sarà presente quindi solo una quantità molto ridotta del fattore VIII nel sangue. A questo punto è necessario somministrare un nuovo fattore affinché la coagulazione possa avvenire efficacemente. Maggiore è quindi l’emivita, minore è la frequenza con cui il paziente deve autosomministrarsi il medicamento. Il video della Società Svizzera Emofilia fornisce una spiegazione chiara dell’emivita.
Gli inibitori ostacolano il trattamento
Nell’ambito della sostituzione di un fattore della coagulazione può verificarsi una grave complicazione, ovvero lo sviluppo di una particolare sostanza, chiamata inibitore, che riduce drasticamente l’azione del fattore sostituito.
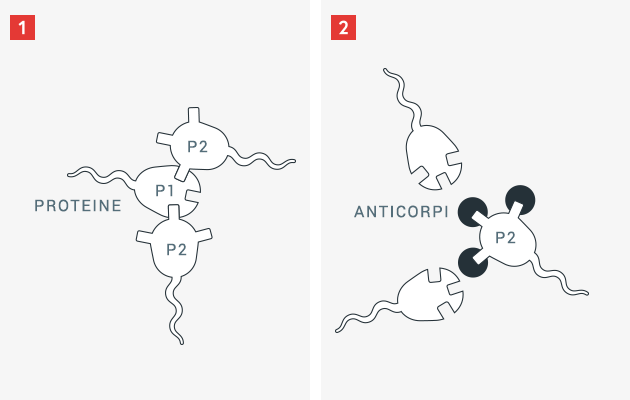
Il sistema immunitario umano è strutturato per garantire protezione contro i pericoli, in particolare contro gli agenti patogeni invasori quali batteri, virus e funghi. Per proteggersi contro gli onnipresenti intrusi, il sistema immunitario ha sviluppato la capacità di distinguere le caratteristiche superficiali estranee dalle strutture e dalle proteine proprie del corpo. Non appena una proteina estranea viene riconosciuta dal sistema immunitario, le cellule immunitarie producono, con una serie di passaggi intermedi, sostanze di difesa, i cosiddetti anticorpi.
Gli anticorpi sono proteine che possono legarsi in modo molto specifico a determinate strutture superficiali. Grazie a questo legame il corpo può rendere inefficace una proteina estranea ed eliminarla più facilmente. Nel caso delle proteine proprie del corpo, ciò non accade, poiché nel corso del loro sviluppo il corpo umano e il suo sistema immunitario imparano la differenza tra «estraneo» e «proprio». Poiché i pazienti con una carenza congenita del fattore della coagulazione non hanno questa proteina, o la hanno solo in forma modificata (e quindi meno efficace), il sistema immunitario non impara a riconoscere come «propria» questa proteina della coagulazione solitamente presente. Se nel paziente viene introdotta la proteina della coagulazione necessaria (ad es. il fattore VIII), può accadere che il sistema immunitario identifichi questa proteina come «estranea» e produca specifici anticorpi per contrastarla.
Nel campo dell’emofilia questi anticorpi sono anche noti come inibitori. Questi anticorpi specifici possono rendere la proteina della coagulazione introdotta inefficace, meno efficace e meno duratura, così che il paziente perde la protezione contro i sanguinamenti acquisita con la somministrazione del medicamento.
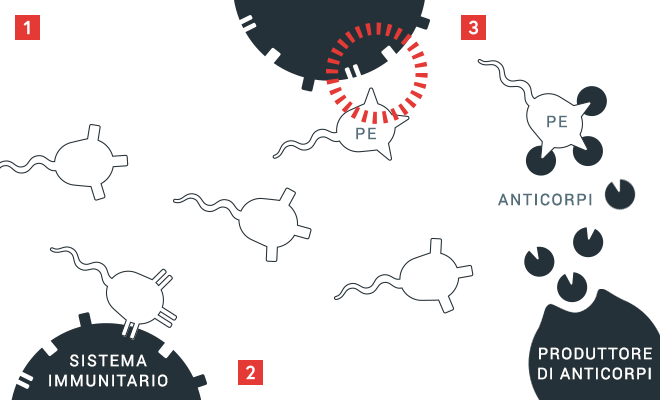
Principio della formazione di inibitori
1 Il sistema immunitario controlla le proteine sulla base delle caratteristiche “proprie” a lui note.
2 Se le proteine vengono riconosciute come «estranee» (PE), vengono prodotti anticorpi
3 che si legano a queste caratteristiche ignote. In caso di formazione di inibitori, vengono prodotti anticorpi contro il fattore VIII iniettato, che diventa di conseguenza inefficace.
Fattori di rischio per la formazione di inibitori
Non tutti i disturbi della coagulazione comportano lo stesso elevato rischio per la formazione di inbitori durante la terapia sostitutiva (sostituzione del fattore mancante). Il rischio della formazione di inibitori è più alto durante le prime 50 somministrazioni; dopodiché, entro la 150ª somministrazione, raggiunge un livello ridotto che rimane stabile.
Il rischio della formazione di inibitori è legato a fattori congeniti, dunque non influenzabili, come ad esempio la provenienza etnica o la presenza di un parente con inibitori, e a fattori di rischio acquisiti, che possono essere ridotti e che sono oggetto della pianificazione della terapia da parte del medico con il paziente. Se gli inibitori sono presenti e il paziente sanguina, occorre continuare a trattare il paziente per evitare danni dovuti al sanguinamento.
Approcci terapeutici
Oggi, in presenza di un disturbo della coagulazione con formazione di inibitori, sono disponibili diverse terapie che permettono di attivare la produzione di fibrina e di riprodurre, quindi, una normale coagulazione del sangue.
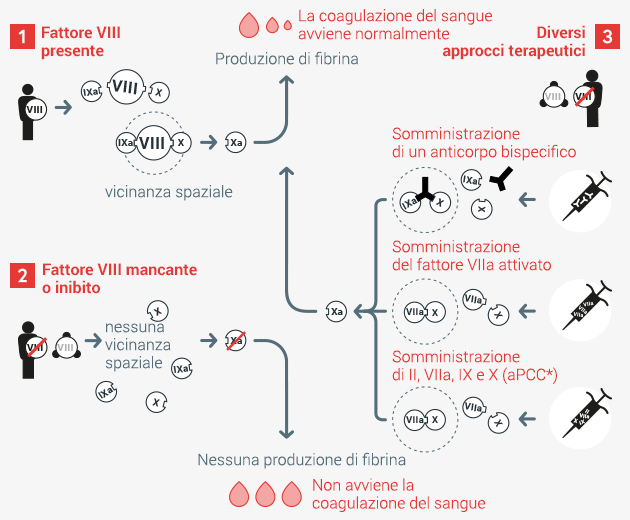
Approcci terapeutici in caso di disturbo della coagulazione del sangue *aPPC = complesso protrombinico concentrato attivato
Caso 1 Nella normale coagulazione del sangue il fattore VIII, in stretta vicinanza con i fattori IX e X, garantisce la formazione del fattore Xa. Quest’ultimo è necessario per la produzione di fibrina che permette la coagulazione del sangue.
Caso 2 Se il fattore VIII manca o non è attivo a causa della formazione di inibitori, viene meno la vicinanza tra i fattori IXa, VIIIa e X. Il fattore Xa non viene prodotto in modo efficace e la coagulazione è quindi inibita.
Caso 3 Somministrando un anticorpo bispecifico, il principio attivo fa avvicinare tra loro i fattori IXa e X, senza aver bisogno del fattore VIII. In alternativa, anche la somministrazione del fattore VIIa attivato o in combinazione con l’aPPC (complesso protrombinico concentrato attivato) può provocare la formazione del fattore VIII e garantire, di conseguenza, una normale coagulazione del sangue.
In tutti e tre i casi il fattore Xa viene prodotto efficacemente e la coagulazione del sangue avviene regolarmente.
È possibile «bandire» nuovamente un inibitore dal corpo, facendo imparare al sistema immunitario, tramite una terapia, a tollerare la proteina della coagulazione introdotta («terapia di tolleranza immunologica»). Questa terapia può durare mesi o anni ed è molto faticosa per il paziente. In determinati pazienti che presentano una particolare azione inibitoria, il sistema immunitario viene ulteriormente soppresso in maniera medicamentosa per impedire la formazione di inibitori.
- Schweizerische Hämophilie-Gesellschaft www.shg.ch
- World Federation of Hemophilia www.wfh.org
- Selpers www.selfers.com
- Deutsche Hämophiliegesellschaft www.dhg.de
- Schmerz Nachrichten OÄ Dr. Waltraud Stromer: Hämophilie und Schmerztherapie (pdf)
- Uniklinikum Saarland Dr. med. Susan Halimeh: Die Bedeutung der Physiotherapie in der modernen Hämoph…